
Drug Development Pathway Costs
Overview
Close to a billion dollars is needed to develop a new drug. However the direct costs for getting a successful drug to market are 100x lower, indicating there is much wasted effort. Motivated entrepreneurs have found much more efficient ways. In this page, we illuminate the stages where cost and waste occur. Then we propose a quick and cost effective approach to get drug development done in Genetic disease.
The Reported Cost
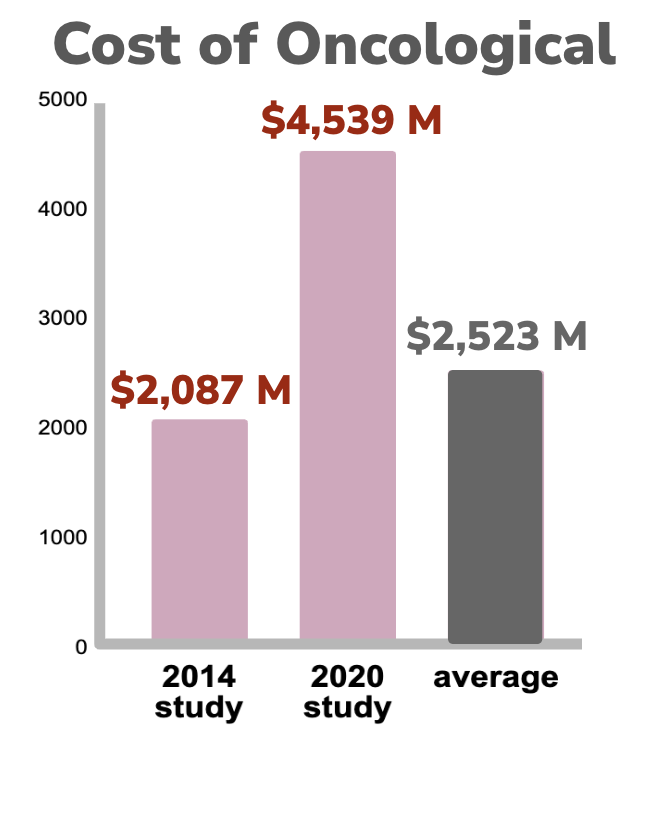
A fully-loaded drug development effort, from target-to-market and including failures, takes about 15 years and costs billions of dollars (PMID: 34368939). Unfortunately these costs are escalating and continue to outpace inflation. Data for Anti-infectives and Neurological drug development indicates the fully loaded costs of getting a drug to market has increased by close to 5x over the last 25 years and is now over $1B for each successful drug (PMIDs 10155302, TIRS, 32125404). In cancer the costs are even higher. The cost of drug to market has increased by more than 2x over a 6 year period and has a high average cost of $2.5B per drug (24419412, 32125404).


The costs of orphan drug comparison vs non-orphan are similar for “out-of-pocket” direct clinical cost (not fully-loaded which does not take into account the challenging preclinical cost of failure and other capital costs) - approximately $ 250 M per drug. However the clinical success rate is 3x higher for orphan drugs, which offsets the 75% higher out of pocket cost observed in getting through clinical trials (PMID 30630499).

Intriguingly, in the above orphan vs non-orphan study, the calculation of the fully-loaded capitalized cost expense is about 4-6x higher (32125404), however the exact fully loaded cost remains controversial (PMID 34368939). The difficulty in estimating the cost is due to the various assumptions in the complex process of drug development. Key driver in calculating correct costs is attempting to determine the cost of failure, which occurs at high frequency in early drug development and with a fairly predictable frequency in clinical trials. However, if we ignore cost of failure for now, we can just look at direct costs estimates for preclinical, which are estimated to be near $7 M (Pharmaceutical Sciences Encyclopedia) and direct costs for clinical development are near $40 M (PMID 26908540) - combined: a total of $47M direct costs. As a result, with median cost for fully-loaded development (cost of failure + capital costs) as being near $1 B, shows that there is quite a bit of inefficiency with the current system (approximately 100-200x wasted effort).

This costing discrepancy and inefficiency makes one wonder if drug development can be done at drastically lower cost?
In a form of an affirmative answer, Terry Pirovolakis, a highly-motivated father of a child with compound heterozygous variants in AP4M1, set out to battle the grim prognosis of SPG50, which is the progressing Spastic Paraplegia 50 that is associated with AP4M1 deficiency. Kids with pathogenic variants in this gene become paralyzed through spasticity and have sever developmental delays. Terry’s family got this diagnosis in 2019 for their son. But they did not resign themselves to this grim fate. Terry formed a charity (CureSPG50) and later a Pharma company, Elpida Therapeutics SPC. And by 2023, Terry’s team was able to publish the key preclinical data showing both safety and efficacy of an AAV treatment in animal models (PMID 36951961 ). This unlocked clinical trials and allowed them to be treating their son at 3 years out from his diagnosis. Terry is now finishing up his combined Phase 1 + 2 study, and Elpida will be entering pivotal trials with the lead that hopefully will enable an NDA application. The calculated expenses from Terry’s team is $ 4.5 M and counting, but they expect to achieve market approval on an New Drug Application (NDA) for under $ 25M, and that will have been done on a complex biologic, he feels it can be done for a lot less on future projects even lower than $10m!
How Did Terry Poirovolakis do it?
Terry realized that taking charge and forming a charity was essential tool to raise the funds needed that would be impossible any other way. This is due to the limited number of patients and no commercial interest from larger organizations. He laid out clear plans with consecutive milestones of what was the fastest possible timelines to get to the incremental steps in finding a drug that is most importantly safe and efficacious. Although steps tend to be considered as gates to the next step, Terry gambled on the certain steps that had long lead times and continued with the next subsequent step so that he could stay on his timeline. Ultimately the extremely rapid concurrent plan panned out and he filed his Health Canada Clinical Trial Applications (CTAs) late 2022 and subsequently a Investigational New Drug (IND) in early 2023. To date over 5 children have been treated with many more being slated for 2024/2025.
Thanks to the collaborative mindset of the FDA and lessons learned from Covid, Terry and the team at Elpida where able to work with the FDA to gain support for a Phase III pivotal study. Terry hopes that the Gene Therapy will show to be efficacious and give children living with this terrible disease a better life. If all goes well and the trial is a success Terry hopes Elpida will be able to obtain a Priority Review Voucher (PRV). PRVs are valuable to a company for future use. Once the original drug development program gets to market with an NDA approval, the PRV can be used to get accelerated NDA review of any other drug. This shaves off 6 months time to approval and, if used on a blockbuster drug, it is potentially worth billions to obtain - think Ozempic with $14 B in sales last year! Intriguingly the PRV can be sold from one company to another company. The current aftermarket price for a PRV is $100 M+. Terry hopes if one of these vouchers can be received it, would allow his Social Purpose Company to take those funds and create treatment for an exponential number of programs that have no commercial interest and hopefully find a feasible path forward for all rare disease programs.
Asset Valuation
Using the FDA submission process as key milestones in drug development, we can run estimates of drug asset cost, timeline, and efficiency at various time points.

It starts with the discovery step in preclinical. This is where drug volume is highest and the per drug cost the lowest. With discovery of target engagement, we can prep paperwork for a preIND meeting. This is where we share with the FDA our finding that suggest efficacy and tell them what we plan to do for safety and manufacturing to ensure that we can get into the clinic with a safe drug. The fail here is quite high - many drugs are screened from high throughput libraries and only a small fraction of these prove to have good and reproducible effects. Next, we enter preclinical development, this where where animal safety studies, Chemistry, Manufacturing and Controls (CMC), and a clinical plan start coming together. Failure here is still quite high and only a few IND get filed. If an IND gets approved we can enter the three clinical phases. First clinical phase (Phase I) is safety and fails about 9 of 10 times tried. Next clinical phase (Phase II0 is sampling for efficacious dose, which fails about a 3 of 4 of times. However good results here alow prparation of a pre-NDA meeting with the FDA. Next we enter the pivotal phase (Phase III ), where we perform clinical trials to test for effectiveness on large population. Phase II fails about 50% the time. At various timepoints along this timeline we can contemplate partnering the drug asset with Pharma to have them help us finance and guide the later stages of drug development.

The above table indicates some of the licensing revenue that can be achieved at various development stages. However, a few assumptions are made in generating this table. First, the per month drug costs to the patient is $833.00 dollars. However, the per month cost in Rare Disease treatments can be much higher. Alternatively, once a drug falls off patent, the market competition can drive down this cost to the patient by an order of magnitude. As a result, there is quite a bit of leeway in estimating the yearly market potential for a drug to treat a group of patients. Another assumption is that the IP-protected market run is 5 years. For the 1000 patient population netting $10 M per year, that is a market exclusivity net of $50 M.
Now we can examine the royalties we can gain from licensing a program to large Pharma. The total royalties from preIND to NDA on a 1000 population size are $1.85 M, which is close to 4% of total market revenue achieved over the 5 years. Regrettably, at the “Direct Cost Per Stage” as listed the “Drug Discovery and Development” figure, only the large population size of 100,000 is profitable to license (see the white labeled numbers of “Milestone Payments” in “Licensing Revenue” figure). If we change our assumption on drug pricing and bump it by more than 10x to a $10,000 spend per month, then the revenue haul increases by order of magnitude, which then allows for a 10-fold increase in milestone payment requests. The result is the 10,000 and the 2000 patient sizes become profitable to license (pink numbers). However the 1000 patient population size remains challenged for profitability (red numbers). It is these high drug costs (>$100,000 per year) that enable treatment in Rare to be potentially profitable. Furthermore, these types of drug pricing and their research cost estimates support the often-shared patient population cut off of 1500 persons as being on the edge of economic feasibility for Pharma.
PRV Impact
The Highly Rare (100s of patient population) and Ultra Rare (10s of patient population) can still have economic viability to develop drugs when they have access to the PRV (see review by RDCC). Using our “Drug Success Rate'' numbers in “Drug Discovery and Development” figure, we can realize the following licensing deal benefits from having a PRV at each of the 4 stages of drug development.

The result is that the PRV valuation always offsets the cost of drug development, no matter how small the population size. Unfortunately, federal policy changes in the USA over the last 5 years have eroded many of the market incentives in rare and threaten to abandon health care access for these patient populations in the Highly Rare and Ultra Rare categories (Health Capital Group). Advocacy on capitol hill is greatly needed!
Entity Types
Much of the costing we have seen for drug development is done with new chemical entities and biologics in mind. For drug-repurposing, the values in the preclinical side are an order of magnitude smaller due the inherent safety of FDA-approved assets. With repurposing, the biggest challenge to profitability is off-label prescribing by clinicians. Yet for the Pediatric Rare Disease community, where PRVs are accessible. The economics can still be quite profitable to move a program toward NDA. More easy to protect and create market exclusivity are the biologics of Antisense Oligonucleotides (ASO) and Adeno-Associated Virus (AAV). As a result, the higher costs of biologics is often economically justified by their better protectability. New Chemical Entities (NCE) are like the biologics in that they are also highly protectable too. However, a major drawback to NCEs and biologics is their long timeline to market approved therapy, thus drug repurposing is a good choice for a starting place when speed-to-access is the most important consideration.
Fast Therapeutic Development
Speed to a therapy is often the top concern for PAGs. The patient has experienced the Diagnostic Odyssey, which often took them years to get accurately diagnosed. The patient then transitions to the Therapeutic Odyssey, where often there is no targeted therapeutic available that will directly treat the disease. As a result, the desire is quite high to quickly find a therapeutic approach. At Devinebio, we advocate for a smart approach that first starts with drug repurposing which allows us to quickly and affordably find candidates. We then transition to the slower to develop (but more IP protectable) assets - however, we gain efficiency because we use the same drug discovery tools used for repurposing to find effective ASOs, AAVs and NCEs. Devinebio recommends this two step approach is best optimized by participating in special drug discovery companies, the “SPECs” who are created for the sole purpose of finding drugs to a gene-specific disease indication. Bottomline, the SPECs will first start on FDA-approved drug assets, but also proceed to trying to find better assets with ASOs, AAVs and NCEs, where appropriate.
Ready to partner on drug discovery?
Schedule a call with us using the following link: